Síndromes de Prader-Willi y de Angelman: aspectos genéticos
Prader-Willi and Angelman syndromes: genetic aspects
Gabriel Márquez, Francisco Montecinos, Francisco Moya
Resumen
Este texto completo es la transcripción editada y revisada de una conferencia dictada en el Curso de Genética, organizado por el Instituto de Ciencias Biomédicas de la Universidad de Chile entre los meses de marzo y julio de 2004.
Edición Científica: Prof. Laura Walker Bozzo, Profesora Encargada de Curso; Prof. Luisa Herrera Cisterna, Coordinadora de Curso.
Descripción y características clínicas
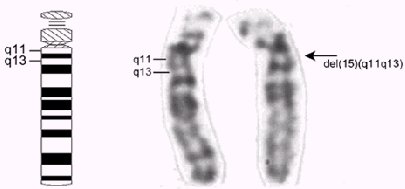
Estos síndromes corresponden a enfermedades multisistémicas complejas que se producen por trastornos genéticos asociados a una alteración (microdeleción, disomía uniparental, mutaciones puntuales o falla en el imprinting) del brazo largo (q) del cromosoma número 15, específicamente de la región 15q11-q13, como se ve en la figura 1. Si el cromosoma que experimenta la alteración es de origen paterno se produce el síndrome de Prader-Willi, mientras que si este cromosoma es de origen materno, se produce el síndrome de Angelman.
Figura 1. A la izquierda, esquema que indica la ubicación de los segmentos del cromosoma 15 que comúnmente se ven afectados en los síndromes de Prader-Willi y de Angelman. A la derecha, par cromosómico número 15 bandeado G con microdeleción en el segmento 15q11-q13, en uno de los cromosomas homólogos (flecha, banda 550).
En el síndrome de Prader Willi (SPW), los individuos afectados presentan hipotonía neonatal, baja estatura, hipogonadismo, retardo mental de grado moderado a severo, apariencia física característica, con diámetro bifrontal reducido, ojos "almendrados", ángulos de la boca dirigidos hacia abajo y manos y pies pequeños en relación a su estatura, e hiperfagia compulsiva que conduce a obesidad precoz y severa.
En el caso de síndrome de Angelman (SA), los síntomas son marcha atáxica, microcefalia, retraso mental, capacidad de habla mínima o nula, con comunicación receptiva y no verbal más desarrollada que la verbal, convulsiones, trastornos del sueño, hiperactividad (que disminuye con la edad) y personalidad con risa frecuente.
Bases genéticas
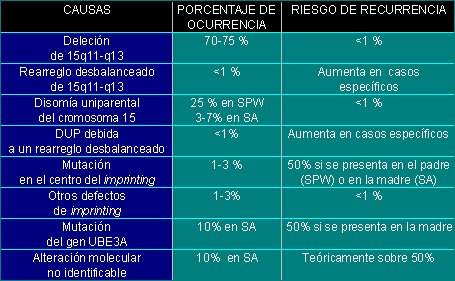
Dependiendo de la causa que genera la enfermedad, el patrón de herencia será diferente y el riesgo de contraer la enfermedad también será distinto, aunque, en general, el tipo de herencia es no mendeliana. Las causas, la incidencia y el riesgo de transmitir la enfermedad a la descendencia (riesgo de recurrencia) se detallan en la tabla I.
Tabla I. Causas, porcentajes de ocurrencia y riesgos de recurrencia de los síndromes de Prader-Willi y de Angelman.
La edad de aparición de ambos síndromes es al momento de nacer, sin embargo, la patogénesis se produce en etapas tempranas del embrión (blastocisto) o durante la gametogénesis de los padres. La expresividad y penetración de estas enfermedades es, en general, completa, pero cuando son causadas por motivos distintos a una deleción cromosómica, como una falla en el imprinting o una disomía uniparental (UPD), presentan expresividades distintas. Así, una alteración en el imprinting va a presentar síntomas más o menos marcados, dependiendo de los genes involucrados.
Por ejemplo, en el SPW se presentan diferentes grados de alteración de la pigmentación y en pacientes con SA pueden presentarse distintos niveles de ataxia. En ambos síndromes, la mayoría de las deleciones son destacablemente homogéneas en extensión, con dos alternativas de quiebre en la región proximal respecto del centrómero y una alternativa en la región distal del cromosoma.
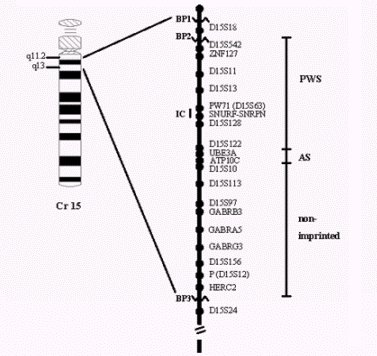
El punto de quiebre proximal de la deleción se encuentra generalmente entre el locus D15S18 y el centrómero (BP1) o entre los loci D15S18 y D15S42 (BP2); el punto de quiebre distal de la deleción ha sido mapeado entre los loci D15S12 y D15S24 (BP3) (véase Figura 2).
Figura 2. Mapa genético de la región 15q-q13 del cromosoma 15 humano. Los puntos de quiebre más comunes del cromosoma, involucrados en la deleción (BP1- BP2 y BP3) se muestran con líneas en zigzag. La barra vertical a la derecha ilustra las regiones críticas para PWS (desde el gen ZNF127 hasta el gen SNRP) y para AS (incluye al gen UBE3A), y la región que no experimenta imprinting. Nótese que las regiones críticas para ambos síndromes incluyen el centro de imprinting (IC).
Se ha encontrado también que, específicamente, la región de deleción causante del SPW está entre los loci D15S13 y D15S10; esta región tiene un tamaño que oscila entre los 100-200 kb e incluye a los genes SNRPN, PAR-5 y PAR-7. En cambio, la deleción causante del SA se encuentra entre los loci D15S122 y D15S113, siendo de un tamaño aproximado de 300 kb e incluyendo a los genes UBE3A y ATP10C. Debido al número y variedad de genes involucrados en cada una de estas deleciones se producirían los diversos síntomas asociados con las enfermedades.
Patogénesis
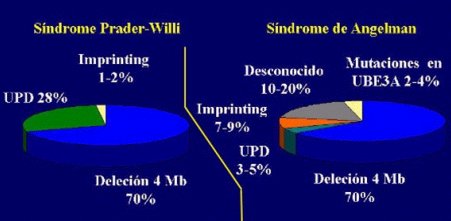
Se describen cuatro alteraciones genéticas principales como causantes de los síndromes; cada una de ellas incide en la aparición de estas patologías, con frecuencias distintas y específicas para cada síndrome (véase Figura 3). A continuación se detallan estas alteraciones.
Figura 3. Frecuencias de las distintas causas que generan los Síndromes de Prader-Willi y de Angelman.
La microdeleción origina aproximadamente el 70% de los casos de ambos síndromes e implica la pérdida de genes debido a una deleción de un segmento cromosómico específico, de aproximadamente 4Mb, en la región 15q11-q13. Esta deleción sería causada por factores externos, como radiaciones ionizantes, rayos UV, radiaciones alfa o por acción de agentes intercalantes, que afectan la síntesis del material genético, aunque la causa específica que provoca la deleción no se conoce.
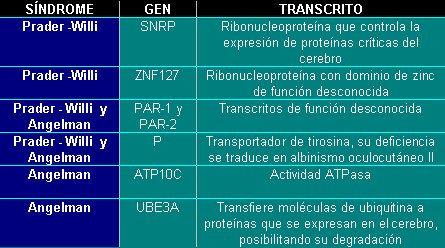
La microdeleción incluye a los genes que producen los síntomas asociados a los síndromes. En la tabla II se muestran los principales genes localizados en el segmento cromosómico que se pierde y sus funciones.
Tabla II. Algunos genes y sus transcritos, involucrados en los síndromes de Prader-Willi (SPW) y de Angelman (SA).
La disomía uniparental (DUP) se produce cuando el individuo afectado, en vez de heredar un cromosoma 15 del padre y el otro de la madre, recibe los dos cromosomas 15 del mismo progenitor. Existen, por lo tanto, dos tipos de DUP; una en la que ambos cromosomas 15 provienen del padre (DUP paterna), en cuyo caso el individuo padecerá de un SA, porque le faltarán los genes activos maternos, y otra en que ambos cromosomas 15 provienen de la madre (DUP materna), y en ese caso el individuo sufrirá de un SPW, porque le faltarán los genes activos del padre.
La DUP materna se relaciona con la ocurrencia de no disyunción durante la meiosis I de la madre, originándose un zigoto trisómico para el cromosoma 15. La letalidad de esta situación se evita gracias a que, por un mecanismo desconocido, se produce un reconocimiento de la falla y se elimina al azar uno de los tres cromosomas 15, fenómeno conocido como "mecanismo de rescate". Una de cada tres veces el cromosoma 15 expulsado es el del padre, originándose una heterodisomía uniparental materna, en la cual un Cr15 procede del abuelo materno del niño y el otro, de la abuela materna.
La DU paterna se produce durante la espermatogénesis; en este caso los dos Cr15 son idénticos, porque ocurre una duplicación del cromosoma una vez que ha segregado normalmente. Esta situación se conoce como isodisomía u homodisomía uniparental, ya que los dos cromosomas 15 proceden del mismo progenitor, que en este caso es el padre. Su origen puede ser una monosomía 15 paterna, por ausencia del cromosoma 15 en el óvulo, debido a un error meiótico en la línea germinal materna, con posterior duplicación compensatoria del 15 paterno. También puede ocurrir por disomía 15 paterna, formación de un zigoto trisómico y expulsión del Cr15 materno por el mecanismo de rescate.
En la falla en la impronta génica (imprinting) se produce una conservación del patrón de metilación de uno de los dos Cr15 en la región q11-q13, porque en las células germinales ocurriría una mutación de los genes localizados en el centro de imprinting (IC) del cromosoma, cuya función es controlar el borrado de la impronta de esa región cromosómica. Al no poder efectuarse el borrado de la impronta y quedar esa región metilada y, por lo tanto, genéticamente inactiva, se produce el síndrome de Prader-Willi si ello ocurre en el cromosoma 15 paterno y el síndrome de Angelman, si sucede en el cromosoma 15 materno.
Finalmente, la mutación del gen UBE3A, que es de baja ocurrencia, es una causa específica de la aparición del síndrome de Angelman. Este gen codifica para una proteína, la E6-AP, cuya función es transferir moléculas de ubiquitina a proteínas que se expresan en determinadas regiones del cerebro, lo que posibilita la degradación de las mismas.
Diagnóstico genético
El diagnóstico de cualquiera de estos dos síndromes se puede realizar mediante varios métodos, o por la combinación de éstos. Los métodos que describiremos en este trabajo son el análisis de metilación, la citogenética clásica, la técnica de FISH y el estudio de microsatélites mediante PCR.
El análisis de metilación se basa en la caracterización y comparación del patrón de metilación del ADN, que es específico según los cromosomas sean de origen paterno o materno. Esta metilación diferencial permite distinguir el origen parental de las regiones 15q11-q13.
La citogenética clásica permite la identificación de grandes deleciones en el segmento 15q11-q13 y otras alteraciones cromosómicas que pudieran ser responsables de fenotipos similares. A pesar de que el estudio citogenético clásico detecta un porcentaje bajo de los casos de SPW/SA, aquellos con deleciones extensas en el segmento 15q11-q13, permite descartar otras alteraciones cromosómicas que pudieran ser relevantes para el consejo genético.
El análisis mediante FISH permite la identificación precisa de las deleciones en el segmento15q11-q13, que serían la causa primaria más frecuente de SPW y de SA. Con esta técnica se pueden detectar, con bastante fiabilidad, deleciones pequeñas al interior del segmento 15q11-q13. Se realiza a partir de sangre del paciente y para la hibridación se utilizan, como sondas, el marcador D15S10, que hibrida con la región crítica del SA que contiene al gen UB3EA y SNRPN, que corresponde a un gen involucrado en la región crítica de SPW.
El estudio de microsatélites (PCR) requiere que se disponga de ADN del paciente y de los padres y marcadores del ADN (microsatélites), cuya distribución es propia del genoma de cada individuo, para seguir la herencia de los cromosomas 15. Esta técnica permite determinar la procedencia de los cromosomas, indicando si el hijo ha recibido un cromosoma 15 de cada progenitor (herencia biparental), o si sólo ha recibido cromosomas 15 paternos (DUP paterna) o maternos (DUP materna).
Incidencia y manejo
Con respecto a la frecuencia alélica en distintas poblaciones, se estima que el SPW tiene una frecuencia de 1 en 12.000 a 1 en 15.000 nacidos vivos y que no existen diferencias entre los sexos ni entre las distintas etnias. El SA se presenta con una frecuencia de 1 en 12.000 a 1 en 20.000 nacidos vivos, no encontrándose tampoco variaciones para los distintos sexos ni razas.
El consejo genético es indispensable, ya que se debe informar a las familias acerca del diagnóstico y pronóstico de la enfermedad y sobre el riesgo de recurrencia del síndrome en la familia, el que depende de la causa que origina el SPW o el SA en el paciente. También se les debe informar sobre la posibilidad de diagnóstico de portadores y de diagnóstico prenatal.
Actualmente, la tendencia del consejo genético es abarcar el mayor número de objetivos posibles, con la finalidad de que un mayor conocimiento permita a la familia tomar decisiones, reducir el estrés sicológico, restaurar los sentimientos de control personal y posibilitar, con el tiempo, una adaptación a la situación.
Detección de Portadores
Se utiliza para detectar individuos que tienen una mutación génica relacionada con la enfermedad, pero que no presentan síntomas. Esto se hace para detectar portadores de Prader- Willi en individuos que tienen algún familiar con el síndrome; por ejemplo, mediante la técnica de FISH pueden detectarse mutaciones en el padre del individuo enfermo, que se pueden transmitir a su descendencia. Si este padre es asintomático, se puede inferir que heredó el defecto de su madre, por lo que también habría que hacer la prueba en ésta.
En el caso del SA, si un individuo presenta el síndrome debido a mutaciones en el gen UBE3A, se recomienda hacer un test a la madre del individuo, realizando FISH con una sonda específica para UBE3A. Con esto se pretende detectar si la madre en cuestión presenta microdeleciones en la zona crítica para Angelman del cromosoma 15. En este caso, la madre podría haber heredado esta mutación de su padre, motivo por el cual ella no presentaría la enfermedad.


Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
No hay comentarios:
Publicar un comentario